什么是金属纳米颗粒?我们为什么要研究金属纳米颗粒?
金属纳米颗粒是尺寸在1-100纳米的金属原子聚集体,比光的波长还小。因其尺寸小,会产生量子限域效应,增加或减少金属原子数目会造成其结构、电子和光学性质的显著改变。因此,与宏观金属材料不同,金属纳米颗粒的尺寸、形貌以及元素分布决定其力学行为、表面吸附、运输、催化活性和光电性质。
比如金纳米颗粒常用于标记生物分子,一方面,形状影响金纳米颗粒的生物分布(图1);另一方面,不同大小形状的金纳米粒子会显现出不同的颜色(图2)。如果想要得到红色的金纳米颗粒,就需要在合成过程中严格控制颗粒的长宽比,否则就很有可能会得到蓝色的颗粒,同时还需要注意不要生成空心的颗粒。又譬如近年来,传统被认为化学“惰”性的金在纳米尺度表现出特殊的催化性能,其尺寸和形貌是决定催化性能的关键因素(图3)。

图1形状影响聚乙二醇修饰的金纳米颗粒在小鼠体内的生物分布,引自Arnida等(2011)

图2结构决定金属纳米颗粒的光学性质,a金纳米棒,引自Dreaden等(2012),b硅核金壳纳米颗粒,引自West等(2003),c银纳米立方体外的金纳米笼,引自Skrabalak等(2007)

图3金纳米颗粒的尺寸和形貌决定其催化性能,在电催化将二氧化碳还原为燃料的过程中,球状Au25(SR)18(上)比棒状Au25(PPh3)10(SR)5Cl2(下)的催化性能更高,Au28(TBBT)20(上)对环己烷和苄醇的有氧氧化过程的催化转化率比Au28(CHT)20(下)更高,在4-硝基酚的还原反应过程中,Au38(PET)24的两个同分异构体Au38T(上右)和Au38Q(上左),前者的催化性能更高,引自Higaki等(2019)
当前金属纳米颗粒的研究难点在哪儿?
直到本世纪初,尺寸均匀性一直是纳米颗粒合成的最大问题,因为它对于系统生产具有均匀物理化学性质的纳米颗粒至关重要。
自从2007年得到Au102(SR)44的晶体合成和表征以来,纳米科学领域在1-3纳米范围内的不同尺寸的金属纳米颗粒的合成方面取得了重大进展。但由于合成方法困难及产量低,如何大规模合成同时具有精确原子数和特定结构的金属纳米颗粒,目前仍是一个挑战性的课题。比如,目前生产的金纳米颗粒有10纳米左右的尺寸分散度(图4)。

图4目前实验手段制备的金纳米颗粒的尺寸分散度在10纳米左右,引自Bailly等(2019)

图5CdSe@Au杂化纳米颗粒的光吸收性能被显著提升,引自Wu等(2015)
金属纳米颗粒是如何“生长”的?
合成金属纳米颗粒最常见的方法是从金属盐的水溶液中制备纳米颗粒。当溶液中溶质的浓度超出溶解度后就会有晶体析出,这就是成核。裸露的金属核活性很高,会吸附更多的金属离子或者其它金属核不断“长大”。有机配体覆盖在金属核表面可以阻止其“长大”,形成可以在溶液中稳定存在的单分散的纳米颗粒。如果配体和金属核的化学相互作用强而且数量又多,就可以合成出尺寸小的纳米颗粒,比如,巯基配体和金的化学相互作用很强,可以生产出1-3纳米的具有精确分子式和化学结构的金纳米颗粒。
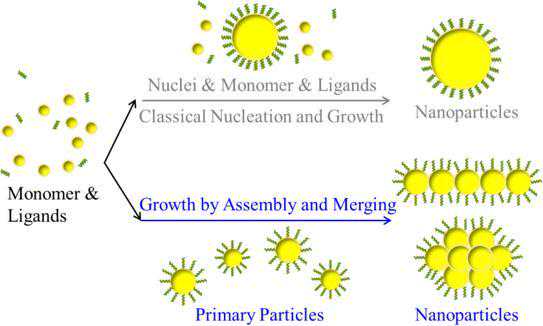
图6金属纳米颗粒的经典成核和生长理论对比融合生长理论的示意图(作图/中科院上海应无所郭盼博士)
金属纳米颗粒的实际成核和“生长”过程非常复杂,自1950年LaMer理论提出至今,科学家在过去的70年里发展了诸多的理论来描述它。我们可以简单的把这些理论分为两类:即金属纳米颗粒增长的单位是“单体”还是“初始颗粒”。
经典的成核理论和晶体“生长模型”认为金属纳米颗粒以单体(金属原子或离子)增加的方式“长大”。但这无法解释合成纳米晶体中观察到的不规则和分枝的晶体形态。基于初始纳米颗粒聚集融合而长成更大的纳米颗粒,这种融合生长模式的提出可以追溯到1973年。2010年代科学家们利用原位液体池透射电子显微镜技术观测到了溶液中金属纳米颗粒的“融合生长”过程。

图7合成金属纳米晶体中观测到的融合“生长”的证据。a铂晶体的融合“生长”形成了铂纳米线,引自Thanh等(2014),b四个二氧化钛晶体通过定向连接融合“生长”形成一个大的晶体,引自Penn等(1999)

图8原位液体池透射电子显微镜下金纳米颗粒的融合“生长”过程,a图上标度尺为2纳米,引自Jin等(2018),b图引自Aabdin等(2014)
为什么要研究金属纳米颗粒的融合“生长”?
融合“生长”为操控金属纳米颗粒的结构提供了巨大的空间。试想,如果晶体以单体增加的方式“生长”,那么最终合成的晶体都趋向于具有无缺陷的规整形状,而如果能够像叠乐高积木一样的把初始晶体叠成更大的晶体,则可以有目的地控制和设计最终晶体的形貌。
比如,科学家可以把配体保护的金属纳米颗粒自组装成纳米颗粒膜,然后再通过加热手段去除自组装膜中相邻纳米颗粒间的配体,从而合成膜状的金属纳米颗粒。科学家还可以通过控制配体的添加量诱导合成金属纳米棒或纳米球。

图9通过融合“生长”方式制备Fe₃O₄纳米颗粒膜,引自Jiao等(2015)

图10通过控制配体的添加量可以利用融合“生长”方式制备不同结构的铂纳米材料,引自Liao等(2013)
了解纳米颗粒的“生长机制”是有目的地设计纳米颗粒的基础。然而受限于目前实验无法观测到融合“生长”过程中纳米颗粒间接触界面的结构变化,包括表面原子、配体以及溶液,融合“生长”的内在物理机制仍有待被阐述。
融合“生长”机制的经典机制
在金属纳米颗粒的合成过程中,配体的保护可以阻止纳米颗粒继续“长大”。在自组装过程中,相邻金属核上的配体以相互交织的状态把金属核隔开,维持自组装结构。配体的作用总是阻碍金属核融合。

图11经典的金属纳米颗粒自组装状态的示意图,引自He等(2010)
经典的金属纳米颗粒的自组装状态非常容易使人产生这样的想法:相邻金属纳米颗粒之间的配体消失后金属核就能融合了。目前普遍认为水溶液中金属纳米颗粒表面配体垂直站立,阻止金属内核接触融合,所以只有颗粒间接触面上的配体完全脱落之后融合“生长”才能发生。

图12目前普遍猜测的金属纳米颗粒融合“生长”过程的示意图
水溶液中配体保护的金纳米颗粒融合“生长”机制的新发现
配体的脱落需要相对较高的温度并且会引起金属内核的严重变形,以巯基配体为例,Au-S键的强度约为50kcal/mol,常温下很难脱落。因此,“颗粒间接触面上的配体完全脱落之后融合“生长”才能发生”的观点无法解释实验中常温下巯基保护的金纳米颗粒的自发融合“生长”。
中科院上海高研院高嶷研究员和郭盼博士通过分子动力学模拟,提出水溶液中巯基羧酸修饰的金纳米颗粒不需要配体脱落的融合“生长”机制。首先水溶液中金纳米颗粒表面配体修饰层的结构是与配体修饰密度相关的。当配体修饰密度高的时候,配体之间的范德华相互作用会促进纳米颗粒表面配体形成结构紧实的自组装层,其中的配体趋向于垂直站立的状态。
而当颗粒表面没有被配体完全覆盖的时候,疏水相互作用促使配体形成结构相对松散的状态,其中配体包裹金纳米颗粒以减少纳米颗粒对于水溶液中氢键网络的破坏。之后配体之间的疏水相互作用主导纳米颗粒形成聚集状态。
处于聚集状态的金纳米颗粒间没有完全被配体覆盖的表面金原子发生接触,融合开始。然后在表面能最小化和金原子之间相互作用最大化的驱使下,接触界面的原子以局部重排的方式把带配体的金原子排出到接触面以外实现完全的融合“生长”。

图13分子动力学方法模拟巯基羧酸修饰的金纳米颗粒的融合“生长”轨迹,引自Guo等(2020)
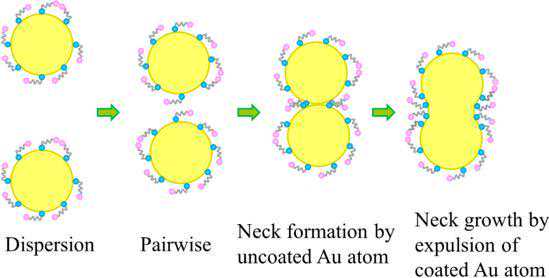
图14水溶液中巯基羧酸修饰的金纳米颗粒的融合“生长”机制示意图,引自Guo等(2020)
该项研究刷新了目前对于金属纳米颗粒融合“生长”原子机制的理解,为今后金属纳米颗粒的合理设计和可控合成奠定了基础。研究成果发表在PhysicalReviewLetter期刊上。
参考文献:
1.Arnida;Janat-Amsbury,;Ray,A.;Peterson,;Ghandehari,H.,Geometryandsurfacecharacteristicsofgoldn,77(3),417-423.
2.Dreaden,;Alkilany,;Huang,X.;Murphy,;El-Sayed,,Thegoldenage:,41(7),2740-2779.
3.West,;Halas,,Engineerednanomaterialsforbiophotonicsapplications:Improvingsensing,imaging,,5,285-292.
4.Skrabalak,;Au,L.;Li,;Xia,,,2(9),2182-2190.
5.Higaki,T.;Li,;Zhao,S.;Li,Q.;Li,;Du,;Yang,S.;Chai,;Jin,,-,58(25),8291-8302.
6.Bailly,;Correard,F.;Popov,A.;Tselikov,G.;Chaspoul,F.;Appay,R.;Al-Kattan,A.;Kabashin,;Braguer,D.;Esteve,,Invivoevaluationofsafety,biodist,9,12890.
7.Wu,K.;Chen,J.;McBride,;Lian,T.,Efficienthot-electro,349(6248),632-635.
8.Thanh,;Maclean,N.;Mahiddine,S.,,114(15),7610-7630.
9.Penn,;Banfield,,Morphologydevelopmentandcrystalgrowthinnanocrystallineaggregatesunderhydrothermalconditions:,63(10),1549-1557.
10.Jin,B.;Sushko,;Liu,Z.;Jin,C.;Tang,R.,InsituliquidcellTEMreveals,18(10),6551-6556.
11.Aabdin,Z.;Lu,J.;Zhu,X.;Anand,U.;Loh,;Su,H.;Mirsaidov,U.,,14(11),6639-6643.
12.Jiao,Y.;Han,D.;Ding,Y.;Zhang,X.;Guo,G.;Hu,J.;Yang,D.;Dong,A.,Fabricationofthree-dimensionallyinterconnect,6,6420.
13.Liao,H.-G.;Zheng,H.,Liquidcelltransmissionelectron,135(13),5038-5043.
14.He,J.;Kanjanaboos,P.;Frazer,;Weis,A.;Lin,;Jaeger,,FabricationandMechanicalPropertiesofLarge‐,6(13),1449-1456.
15.Guo,P.;Gao,Y.,,124(6),066101.
版权声明:本站所有作品(图文、音视频)均由用户自行上传分享,仅供网友学习交流,不声明或保证其内容的正确性,如发现本站有涉嫌抄袭侵权/违法违规的内容。请举报,一经查实,本站将立刻删除。